各行が遺伝子名、各列がサンプル名からなる遺伝子発現行列を視覚化する手法の一つとしてヒートマップを用いる方法がある。R では、ヒートマップを描く関数として、標準的な
heatmap や heatmap.2 の関数の他に、生物学に特化した regHeatmap などの関数が用意されている。
heatmap
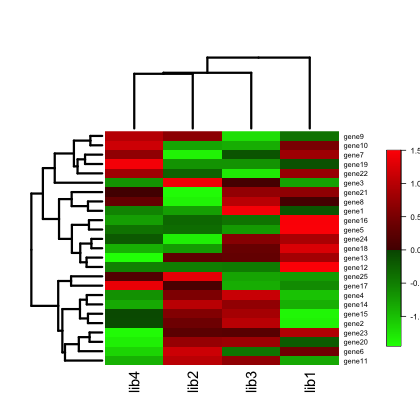
行列型のデータを heatmap 関数に与えると、そのままヒートマップとして描かれる。行と列は、自動的にクラスタリングが行われる。
x <- matrix(rnorm(100), ncol = 4)
colnames(x) <- paste0("lib", 1:ncol(x))
rownames(x) <- paste0("gene", 1:nrow(x))
head(x)
## lib1 lib2 lib3 lib4
## gene1 -0.4752599 -1.0358687 1.0474508 -0.8438768
## gene2 -2.0747126 0.1360623 0.8924237 -0.4413562
## gene3 0.9027044 1.8960928 1.2700205 0.9531910
## gene4 -0.7374487 0.8610647 1.4687483 -0.3636694
## gene5 1.2868677 -0.7670078 -1.1653529 -0.8196983
## gene6 -0.1245493 0.5495175 -0.9170723 -1.5751826
heatmap(x)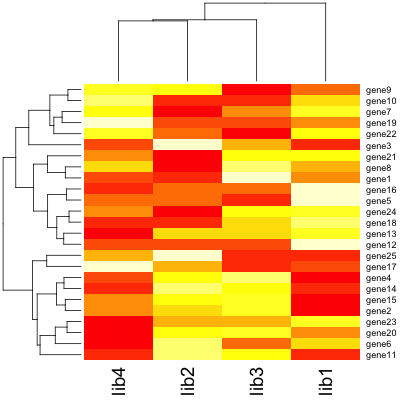
ヒートマップを描くとき、自動的にクラスタリングを行わないようにするには、Colv =
NA または Rowv = NA、または両方を指定すればよい。次は、行をクラスタリングするが、列をクラスタリングしない例である。
heatmap(x, Colv = NA)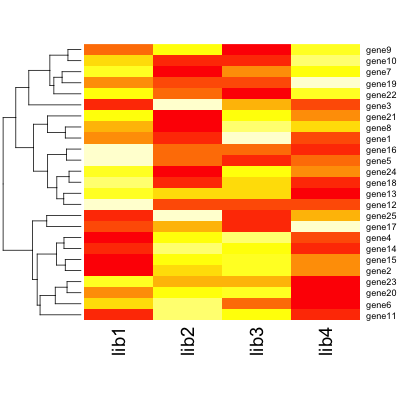
クラスタリングは、デフォルトで dist 関数によって距離が計算されて、 hclust によってクラスタリングが行われる。例えば、hclust
のクラスタリング手法などを変更する場合は、次のようにする。
heatmap(x, hclustfun = function(x) { hclust(x, method = "ward") })heatmap.2
ヒートマップに加えて、密度データやスケールバーなどの付加情報も描きいれる場合は、gplots パッケージの heatmap.2 関数が便利。
library(gplots)
heatmap.2(x)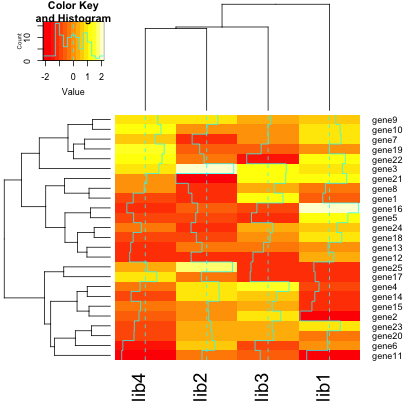
行や列のクラスタリング可否については、heatmap 関数と同様に Colv = NA や Rowv = NA なで指定する。
regHeatmap
Heatplus に含まれている regHeatmap 関数もヒートマップを描くことができる。Heatplus パッケージは Bioconductor に含まれているので、次の方法でインストールする必要がある。
source("http://bioconductor.org/biocLite.R")
biocLite("Heatplus")heatplus を利用してヒートマップを描く例。
library(Heatplus)
regHM <- regHeatmap(x)
plot(regHM)