遺伝子の発現量行列を利用してネットワーク解析の一例を示す。ここでは発現量をそのままを利用しているが、目的に応じて、fold-change に変更や何らかのアルゴリズムで変換したデータを用いることも可能である。
ネットワーク解析においてサンプルが多ければ多いほどがよい。ここで利用するサンプルデータは ReCount データベースにある組織発現量データを利用する。
遺伝子数が多いため、ここでは平均発現量の高い遺伝子 100 個に対してネットワークを描く。データの正規化なども行う必要はあるが、ここでは省略する。
counts <- read.table("http://bowtie-bio.sourceforge.net/recount/countTables/wang_count_table.txt", row.names = 1, header = TRUE)
counts <- counts[rank(- rowMeans(counts)) <= 100, ]
counts.log <- log10(counts + 1)
dim(counts.log)
## [1] 100 22次に、遺伝子同士の相関係数を計算する。
cc <- cor(t(counts.log))ここで計算した相関係数からなる行列を igraph を利用するためのデータの形に変更する。
library(reshape2)
cc[upper.tri(cc)] <- NA
diag(cc) <- NA
cc.df <- melt(cc)
cc.df <- cc.df[!is.na(cc.df[,3]), ]
head(cc.df)
## Var1 Var2 value
## 2 ENSG00000008441 ENSG00000007237 0.7539953
## 3 ENSG00000034510 ENSG00000007237 -0.3951741
## 4 ENSG00000063180 ENSG00000007237 0.6987645
## 5 ENSG00000067225 ENSG00000007237 0.1476872
## 6 ENSG00000072778 ENSG00000007237 -0.0879947
## 7 ENSG00000074181 ENSG00000007237 -0.3519994データが上のような形になればよい。基本的に 1 列目に起点と 2 列目に終点の遺伝子名があればよい。3 列目の相関係数があってもなくてもよい。ここで、相関係数の絶対値が 0.75 以上の値に対して枝を引くという作業をするので、相関係数を計算している。
library(igraph)
cc.df.sig <- cc.df[abs(cc.df[, 3]) > 0.75, ]
g <- graph.data.frame(cc.df.sig[, 1:2], directed = F)
num.of.v <- length(V(g))
V(g)$size <- rep(5, num.of.v)
V(g)$color <- rep("#E41A1C", num.of.v)
V(g)$shape <- rep("circle", num.of.v)
V(g)$label <- names(as.list(V(g)))
V(g)$label.cex <- rep(0.5, num.of.v)
V(g)$label.color <- rep("black", num.of.v)
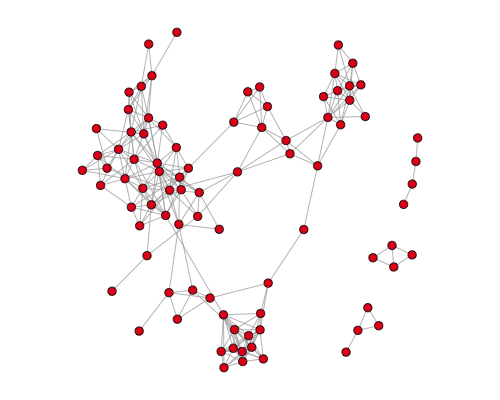
plot(g)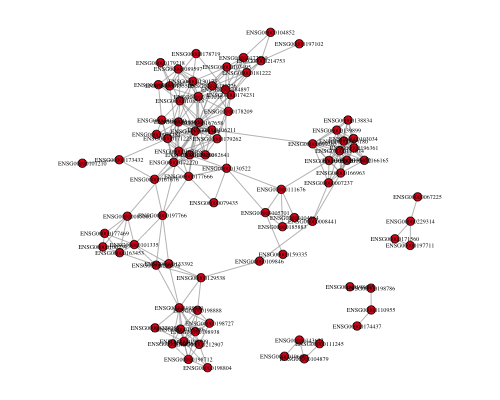
plot(g, vertex.label = NA)