系統樹のデータは、newick フォーマットあるいは nexus フォーマットによって記載される。R では、ape や ade4 などのパッケージを利用することで、これらのフォーマットのファイルを扱うことができる。
ファイルフォーマット
newick フォーマット
newick フォーマットは、括弧を利用して、近い 2 つの要素を次々と囲んで行くフォーマットである。1 例を示すと以下のようになる。括弧の順序に着目すると、A と B はまず 1 つのグループに分けられ、その次に C と D が次々と加わって行く。また、系統樹に距離が定義されているとき、現在の要素とその上の要素の距離を、その要素の横に書く。
((A:0.3,B:0.2):0.4,C:0.6,D:0.8);
nexus フォーマット
上と同じデータを nexus フォーマットで示すと、次のようになる。
#NEXUS [R-package APE, Sat Dec 2 15:08:28 2017] BEGIN TAXA; DIMENSIONS NTAX = 4; TAXLABELS A B C D ; END; BEGIN TREES; TRANSLATE 1 A, 2 B, 3 C, 4 D ; TREE * UNTITLED = [&U] ((1:0.3,2:0.2):0.4,3:0.6,4:0.8); END;
ade4
ade4 パッケージは newick フォーマットの読み込みをサポートしている。newick2phylog 関数は newick フォーマットの文字列を読む関数であるので、もし、ファイルからデータを読む場合は、先にファイルの内容を文字列として読み込んでから newick2phylog 関数に渡す必要がある。
library(ade4)
tree <- newick2phylog("(A:0.3,B:0.2):0.4,C:0.6,D:0.8);")
fh <- file("https://stats.biopapyrus.jp/data/species_tree.txt", "r")
tree <- newick2phylog(readLines(fh, 1))newick2phylog 関数から返されるオブジェクトをそのまま plot 関数に返すと、系統樹が描かれる。cleaves 引数は、系統樹のラベルの近くの黒丸のサイズを指定するための引数である。また、cnodes 引数は、系統樹のノードのところの白丸のサイズを指定するための引数である。
plot(tree, cleaves = 2, cnodes = 2)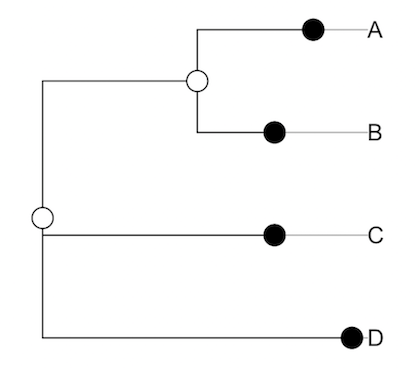
ape
ape パッケージでは、ファイルからデータを読み込むとき read.tree 関数を利用する。また、read.tree 関数が返したオブジェクトをそのまま plot 関数に与えると、系統樹が作成される。
library(ape)
tree <- read.tree("https://stats.biopapyrus.jp/data/species_tree.txt")
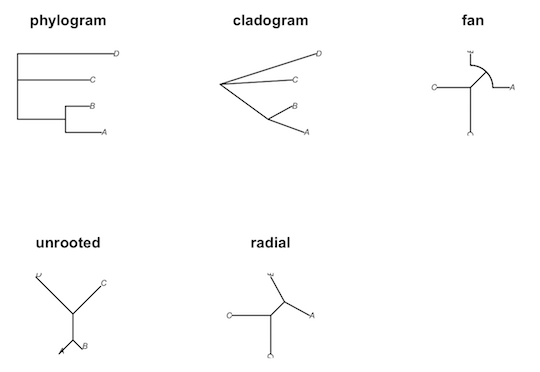
plot(
tree, # read.treeで読み込んだデータ
type = "phylogram", # phylogram, cladogram, fan, unrooted, radial を指定することができる
use.edge.length = TRUE, # FALSE を指定すると、枝の長さは距離情報を含まなくなる!
show.tip.label = TRUE, # 葉のラベルを表示する
show.node.label = TRUE, # ノードのラベルを表示
edge.color = "black", # 枝の色
edge.width = 1, # 枝の太さ
edge.lty = 1, # 枝の種類(実線、点線など)
root.edge = TRUE, # 根を表示する
tip.color = "black" # 葉の色
)type 引数に系統樹のタイプを指定する。各タイプは次のようになっている。
par(mfrow = c(2, 3))
for (type in c("phylogram", "cladogram", "fan", "unrooted", "radial")) {
plot(tree, type = type, main = type)
}
エッジラベル
各枝にラベルを付ける場合は edgelabels を利用する。次は、各枝に、tree$edge.length に保存されている距離情報を付ける例である。
# Sample 1
plot(tree, main = "Sample 1")
edgelabels(
text = tree$edge.length,
frame = "none",
bg = "none",
col = "darkgray",
adj = c(0, 1.2)
)
# Sample 2
plot(tree, main = "Sample 2")
lastEdgeLabel <- tree$edge.length * as.numeric(tree$edge[,1] > tree$edge[,2])
lastEdgeLabel <- ifelse(lastEdgeLabel == 0, NA, lastEdgeLabel)
lastEdgeLabel[1] <- tree$edge.length[1]
edgelabels(lastEdgeLabel, frame="none", bg="none")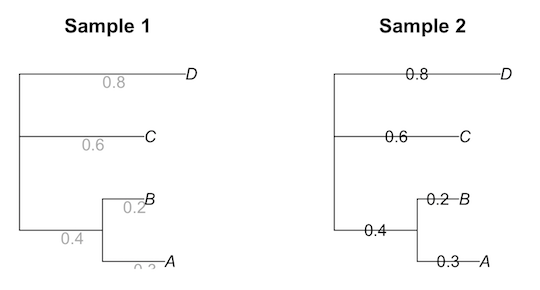
ノードラベル
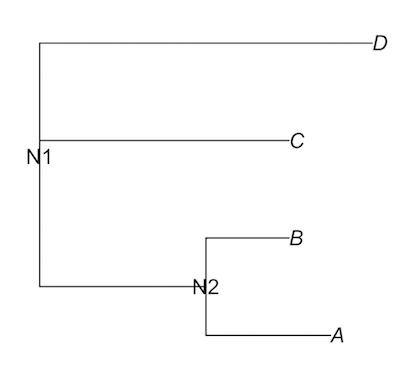
各ノードにラベルをつける場合は nodelabels を利用する。
plot(tree)
nodelabels(
text = c("N1", "N2"),
frame = "none", # デフォルトでは"rect"となり、長方形の枠線が書かれる
bg = "none", # 背景色、デフォルトは緑色になる
col = "black" # ラベル文字の色
)
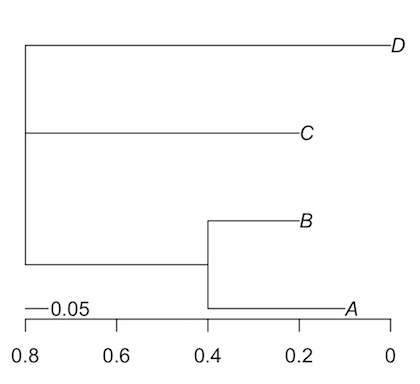
目盛りスケール
横軸に目盛りを付ける場合は axisPhlyo を利用する。また、横軸の目盛りの他に、スケールバーを付け加える場合は add.scale.bar を利用する。
# ノードが右上がりになるようにソート(逆はright = FALSEを指定)
tree.sort <- ladderize(tree, right = TRUE)
plot(tree.sort, type = "phylogram")
axisPhylo()
add.scale.bar(length=0.05)