希薄化曲線は多様性の解析などに用いられる。免疫学でも、T 細胞レセプターにおける V, D, J 遺伝子の組み合わせ数の推定などの利用される。
ここでは実データがないので、乱数を利用してサンプルデータを生成し、データフレーム型でまとめる。このデータを利用して希薄化曲線を描く。
dat.id <- paste0("SeqID_", 1:9900)
dat.d <- sample(paste0("D", 1:3), 9900, prob = c(0.6, 0.3, 0.1), replace = T)
dat.j <- sample(paste0("J", 1:9), 9900, prob = c(0.3, 0.2, 0.14, 0.1, 0.1, 0.05, 0.05, 0.05, 0.01), replace = T)
dat.v <- sample(paste0("V", 1:20), 9900, prob = c(rep(0.05, 10), 0.1, 0.1, 0.02, 0.03, 0.06, 0.01, 0.01, 0.1, 0.065, 0.005), replace = T)
dat <- data.frame(id = dat.id, V = dat.v, D = dat.d, J = dat.j)
dat.size <- nrow(dat)次に、希薄化曲線を描くための座標の初期値を設定しておく。サブサンプリングのサイズが小さいとき、サブサンプリング間隔を小さめにするとよい。(サンプル数が少し上がっただけで、多様性が激増するため。)
x <- c(0, 200, 400, 600, 800, 1000, 1500, 2000, 2500, 3000, 4000, 6000, 8000, 9900)
y <- c(0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0)次に、y 座標を計算する。まず、母集団 dat からサンプル数 x だけサンプリングし、その VDJ の組み合わせを数を計算する。この作業を 1000 回繰り返す(N <- 1000)。このサブサンプリング作業を 1 回だけ行ってもよいが、特に x が小さい時に、サンプリングする度に y 値が大きく変化してしまう。そのため、サブサンプリングの回数を増やして、その平均値をとることで、y のガタつきを減らせる。
for (i in 1:length(x)) {
N <- 1000
boot.values <- rep(0, N)
for (j in 2:N) {
sampled.idx <- sample(1:dat.size, x[i], replace = FALSE)
sampled.dat <- dat[sampled.idx, ]
sampled.vdj <- apply(sampled.dat[, 2:4], 1, paste0, collapse = "_")
sampled.vdj.unique <- unique(sampled.vdj)
boot.values[j] <- length(sampled.vdj.unique)
}
y[i] <- mean(boot.values)
}plot 関数で枠を作成し、lines で線を描き、points で点を描く。
plot(x, y, type = "n")
grid()
lines(x, y)
points(x, y)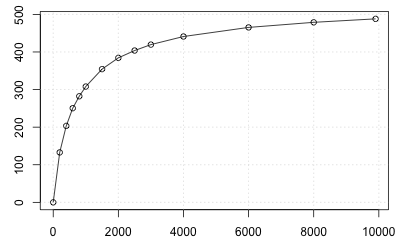
複数の曲線を描く際に、それぞれのデータについて座標を求めればよい。
dat.1.id <- paste0("G1_ID_", 1:9900)
dat.1.d <- sample(paste0("D", 1:3), 9900, prob = c(0.6, 0.3, 0.1), replace = T)
dat.1.j <- sample(paste0("J", 1:9), 9900, prob = c(0.3, 0.2, 0.14, 0.1, 0.1, 0.05, 0.05, 0.05, 0.01), replace = T)
dat.1.v <- sample(paste0("V", 1:20), 9900, prob = c(rep(0.05, 10), 0.1, 0.1, 0.02, 0.03, 0.06, 0.01, 0.01, 0.1, 0.065, 0.005), replace = T)
dat.1 <- data.frame(id = dat.1.id, V = dat.1.v, D = dat.1.d, J = dat.1.j)
dat.1.size <- nrow(dat.1)
dat.2.id <- paste0("G2_ID_", 1:5594)
dat.2.d <- sample(paste0("D", 1:3), 5594, prob = c(0.7, 0.1, 0.2), replace = T)
dat.2.j <- sample(paste0("J", 1:9), 5594, prob = c(0.3, 0.2, 0.1, 0.1, 0.1, 0.09, 0.05, 0.06, 0.00), replace = T)
dat.2.v <- sample(paste0("V", 1:20), 5594, prob = c(rep(0.05, 10), 0.19, 0.01, 0.01, 0.04, 0.06, 0, 0.02, 0.1, 0.07, 0), replace = T)
dat.2 <- data.frame(id = dat.2.id, V = dat.2.v, D = dat.2.d, J = dat.2.j)
dat.2.size <- nrow(dat.2)
# x 座標
x.1 <- c(0, 200, 400, 600, 800, 1000, 1500, 2000, 2500, 3000, 4000, 6000, 8000, 9900)
x.2 <- c(0, 200, 400, 600, 800, 1000, 1500, 2000, 2500, 3000, 4000, 5594)
# y 座標の計算を関数として定義
calcY <- function(d, x) {
d.size <- nrow(d)
y <- rep(0, length(x))
for (i in 1:length(x)) {
N <- 100
boot.values <- rep(0, N)
for (j in 2:N) {
sampled.idx <- sample(1:d.size, x[i], replace = FALSE)
sampled.dat <- d[sampled.idx, ]
sampled.vdj <- apply(sampled.dat[, 2:4], 1, paste0, collapse = "_")
sampled.vdj.unique <- unique(sampled.vdj)
boot.values[j] <- length(sampled.vdj.unique)
}
y[i] <- mean(boot.values)
}
y
}
# y 座標の計算
y.1 <- calcY(dat.1, x.1)
y.2 <- calcY(dat.2, x.2)
# プロット
plot(c(x.1, x.2), c(y.1, y.2), type = "n")
grid()
lines( x.1, y.1, col = "orange", lty = 1)
points(x.1, y.1, col = "orange", pch = 19)
lines( x.2, y.2, col = "darkgreen", lty = 1)
points(x.2, y.2, col = "darkgreen", pch = 15)
legend("bottomright", legend = c("G1", "G2"), col = c("orange", "darkgreen"), pch = c(19, 15), lty = c(1, 1))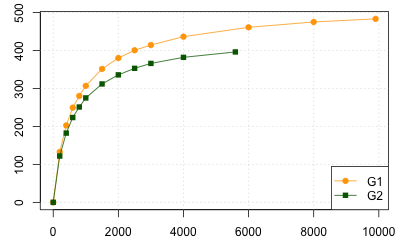
有効数字で表したい場合は sfsmisc パッケージの eaxis を利用すると便利。
library(sfsmisc)
plot(c(x.1, x.2), c(y.1, y.2), type = "n", xaxt = "n") # x軸を非表示にする
eaxis(1, labels = pretty10exp(axTicks(1))) # 1: x軸; 2: y軸
grid()
lines( x.1, y.1, col = "orange", lty = 1)
points(x.1, y.1, col = "orange", pch = 19)
lines( x.2, y.2, col = "darkgreen", lty = 1)
points(x.2, y.2, col = "darkgreen", pch = 15)
legend("bottomright", legend = c("G1", "G2"), col = c("orange", "darkgreen"), pch = c(19, 15), lty = c(1, 1))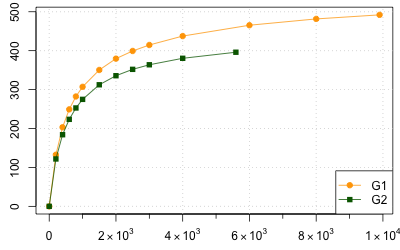
References
- Comprehensive analysis of the T-cell receptor beta chain gene in rhesus monkey by high throughput sequencing. Sci Rep. 2015. 5:10092. PubMed Abstract